 W
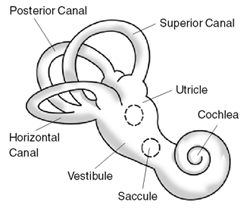
WŁagodne napadowe pozycyjne zawroty głowy – choroba wynikająca z zaburzeń w uchu wewnętrznym. Objawami są powtarzające się, krótkie zawroty głowy, wywołane przez zmianę położenia głowy. Zawroty głowy mogą nastąpić przy skrętach głowy w łóżku lub innych zmianach pozycji. Każdy epizod zawrotów głowy zwykle trwa nie dłużej niż minutę. Zazwyczaj tym zawrotom towarzyszą nudności. ŁPZG jest jedną z najczęstszych przyczyn zawrotów głowy.
 W
WChoroba Ménière’a – rzadka choroba, której przyczyną jest nadmierne gromadzenie się i wzrost ciśnienia endolimfy w błędniku. Objawia się układowymi zawrotami głowy, szumem usznym i postępującą utratą słuchu.
 W
WZespół Duane’a – rzadka wrodzona choroba związana z zaburzeniami w ruchach gałek ocznych. Wiążą się one z nieprawidłowym rozwojem nerwu odwodzącego. Choroba występuje z częstością 1:10000. 1% do 5% osób z zezem wykazuje cechy zespołu Duane’a.
 W
WDystrofia Fuchsa – powoli postępująca choroba rogówki, która zazwyczaj zajmuje oboje oczu i występuje nieznacznie częściej u kobiet. Opisana po raz pierwszy przez Ernsta Fuchsa.
 W
WEktropion, ektropium – odwinięcie powieki. Występuje u ludzi, a także jako dziedziczna przypadłość dotykająca psy dużych ras, mogąca spowodować długotrwałe drażnienie rogówki, w wyniku czego może dojść do jej uszkodzenia. Podobnie skutki powoduje entropion.
 W
WNerwiakowłókniakowatość typu 1 – choroba genetyczna o dziedziczeniu autosomalnym dominującym, należąca do grupy fakomatoz. W obrazie klinicznym choroby występują zmiany skórne, oczne, guzy wewnątrzczaszkowe i inne nowotwory o lokalizacji pozaczaszkowej, a także zmiany kostne. Ze względu na dużą zmienność objawów klinicznych nierzadko rozpoznanie choroby jest opóźnione w przypadkach o łagodnej ekspresji. Nerwiakowłókniakowatość typu 1 spowodowana jest mutacją w genie NF1 kodującym neurofibrominę 1. Choroba ta jest nieuleczalna.
 W
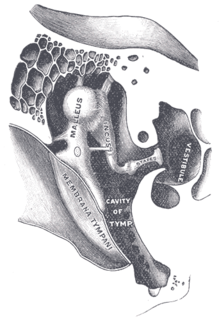
WOtoskleroza – choroba kosteczek ucha środkowego, która stopniowo prowadzi do utraty słuchu. Dolegliwość ta występuje rodzinnie; częściej dotyka kobiety między 15 a 30 rokiem życia. Może znacznie pogłębić się w czasie ciąży.
 W
WReaktywne zapalenie stawów – zespół objawów chorobowych występujących po zapaleniu cewki moczowej lub jelit, na który składają się następujące objawy:zapalenie spojówek lub tęczówki zapalenie cewki moczowej zapalenie stawów – często asymetryczne wędrujące zapalenie kilku stawów, głównie kończyn dolnych, np. stawu kolanowego i skokowego. U niektórych zajęte są też stawy palców rąk i nóg.
 W
WRopostek – obecność ropy w komorze przedniej oka, zwykle towarzyszy mu zaczerwienienie spojówki i leżącej pod nią nadtwardówki. Jest to objaw zapalenia tęczówki i ciałka rzęskowego. Wysięk gromadzi się na dole z powodu grawitacji.
 W
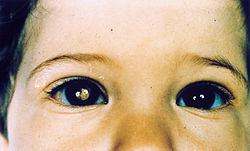
WSiatkówczak – najczęstszy wewnątrzgałkowy nowotwór złośliwy oka u dzieci. Jeśli weźmie się pod uwagę wszystkie grupy wiekowe, zajmuje on drugie miejsce po czerniaku naczyniówki w tej grupie nowotworów. Rozwój nowotworu jest inicjowany przez mutacje, które dezaktywują obie kopie genu RB1, kodującego białko retinoblastoma. Pierwszym objawem choroby jest najczęściej leukokoria – pojawienie się białego odblasku w oku lub obu oczach, albo zez. Siatkówczak jest rzadką chorobą – ocenia się, że zapadalność na ten typ nowotworu wynosi około 4 przypadków na milion na rok.
 W
WStwardnienie guzowate, choroba Bourneville’a-Pringle’a, TSC – rzadka, wielonarządowa choroba uwarunkowana genetycznie, należąca do grupy fakomatoz, powodująca zmiany w nerkach, sercu, gałkach ocznych, mózgu, płucach i skórze, często związana z niepełnosprawnością intelektualną i padaczką. Stwardnienie guzowate zostało opisane jako nowa jednostka chorobowa w 1880 roku przez Désiréa-Magloire’a Bourneville’a. Przez ostatnie dwa dziesięciolecia dokonał się znaczny postęp w wiedzy o tej chorobie, związany z odkryciem jej genetycznego podłoża – mutacji w genach TSC1 i TSC2. Stwarza on nowe możliwości skutecznej terapii stwardnienia guzowatego.
 W
WŚlepota – całkowite lub znaczne zaburzenie widzenia. Ślepota może być spowodowana wadami oka lub chorobami i uszkodzeniami natury neurologicznej lub mechanicznej. Zaburzenie to może mieć charakter nabyty lub wrodzony. Na świecie żyje około 45 milionów ludzi niewidomych i 269 milionów z zaburzeniami widzenia.
 W
WŚlepota zmierzchowa – wada wzroku, polegająca na zaburzeniu widzenia w warunkach słabego oświetlenia. Jest jednym z początkowych objawów retinopatii barwnikowej. Powstaje ona wskutek upośledzenia czynności pręcików w siatkówce oka. Osoby dotknięte ślepotą zmierzchową nie widzą przy słabym oświetleniu. Większość ptaków ma upośledzenie widzenia w warunkach słabego oświetlenia i stąd potoczna nazwa wady.
 W
WZespół Axenfelda-Riegera – zespół wad wrodzonych, charakteryzujący się występowaniem obustronnych wad przedniego odcinka oka skojarzonych z wadami rozwojowymi zębów, środkowej części twarzy i jamy brzusznej. Większość przypadków zespołu wiąże się z mutacjami genów PITX2 lub FOXC1. Zespół opisali niezależnie od siebie Theodor Axenfeld i Herwigh Rieger.
 W
WZespół Harboyana – zwyrodnieniowa choroba rogówki określana też jako wrodzona dziedziczna dystrofia endotelialna ; ponadto, towarzyszy jej postępująca, postlingwalna czuciowo-nerwowa utrata słuchu. Do 2008 roku opisano 24 przypadki choroby z 11 rodzin o różnym pochodzeniu etnicznym. W ponad 50% opisanych przypadków stwierdzono pokrewieństwo rodziców. Objawy oczne w zespole Harboyana obejmują obustronny rozsiany obrzęk rogówki z ciężkim zmętnieniem rogówki, spadkiem ostrości widzenia, utratą wzroku i oczopląsem. Objawy widoczne są po urodzeniu albo w okresie noworodkowym i są nierozróżnialne od autosomalnej recesywnej wrodzonej dziedzicznej dystrofii endotelialnej (CHED2). Niedobór słuchu w zespole Harboyana wolno postępuje i zazwyczaj rozpoznawany jest u pacjentów w wieku 10-25 lat. Nie opisano dotąd przypadków prelingwalnej głuchoty u pacjentów z zespołem Harboyana, jednak w badaniach audiometrycznych stwierdzano znaczącą utratę słuchu już w wieku 4 lat. Choroba wiąże się z mutacjami w genie SLC4A11 w locus 20p13-p12, tym samym co locus genu CHED2: tym samym, zespół Harboyana i CHED2 są schorzeniami allelicznymi. Do tej pory zidentyfikowano 62 różne mutacje genu SLC4A11 u 98 rodzin. We wszystkich tych przypadkach stwierdzono dziedziczenie autosomalne recesywne. Rozpoznanie choroby opiera się o kryteria kliniczne, szczegółowe badanie okulistyczne i badanie audiometryczne. Możliwe jest potwierdzenie zespołu testem genetycznym. W diagnostyce różnicowej zespołu należy uwzględnić szereg chorób genetycznych, metabolicznych, zaburzeń rozwojowych i nabytych uszkodzeń charakteryzujących się mętnieniem rogówki: anomalię Petersa, sclerocornea, dermoid rąbka, zaćmę wrodzoną. W celu zróżnicowania CHED2 z zespołem Harboyana niezbędna jest audiometria. Ponadto, autosomalne recesywne postaci dystrofii rogówki powinny być różnicowane z, charakteryzującą się lżejszym przebiegiem, autosomalną dominującą CHED1. Nieprawidłowości oczne w zespole Harboyana można leczyć hiperosmotycznymi roztworami podawanymi miejscowo w kroplach. Ostatecznym leczeniem jest przeszczep rogówki.
 W
WZespół von Hippla-Lindaua, choroba von Hippla-Lindaua, naczyniakowatość siatkówkowo-móżdżkowa, HLS, VHL – rzadkie schorzenie genetyczne należące do grupy fakomatoz, dziedziczone autosomalnie dominująco. Zespół opisali niezależnie od siebie Eugen von Hippel w 1894 i Arvid Lindau w 1926 roku. Przyczyną choroby jest mutacja w obydwu allelach genu VHL, z których jedna jest dziedziczna a druga następuje spontanicznie. Choroba charakteryzuje się zwiększoną predyspozycją do nowotworów nerek, ośrodkowego układu nerwowego, szczególnie móżdżku, nadnerczy i siatkówki. Nie ma metody leczenia przyczynowego, ale poradnictwo i badania genetyczne pozwalają coraz częściej zdiagnozować ją, zanim pojawią się zagrażające życiu guzy nowotworowe.