 W
WDziedziczenie autosomalne recesywne – w genetyce sposób dziedziczenia, w którym cecha dziedziczona jest w sprzężeniu z chromosomami innymi niż chromosomy płci i ujawnia się tylko w układzie homozygotycznym recesywnym, co oznacza, że obydwa allele genu muszą kodować daną cechę.
 W
WAcyduria mewalonianowa – choroba metaboliczna uwarunkowana genetycznie. Spowodowana jest niedoborem enzymu kinazy mewalonianowej. Objawia się opóźnieniem umysłowym, postępującą ataksją, przełomami gorączkowymi z biegunką i wymiotami, wysypką na skórze, hepatosplenomegalią, cechami dysmorficznymi twarzy, nieprawidłowym rozwojem i wzrostem.
 W
WAlfa-mannozydoza – uwarunkowana genetycznie choroba z grupy lizosomalnych chorób spichrzeniowych, charakteryzująca się pierwotnym niedoborem odporności, nieprawidłowościami szkieletu i cechami dysmorficznymi twarzy, upośledzeniem słuchu i opóźnieniem umysłowym. Chorobę opisał Ockerman w 1967 roku.
 W
WAtaksja Friedreicha, choroba Friedreicha, bezład Friedreicha – choroba o podłożu genetycznym, dziedziczona w sposób autosomalnie recesywny, prowadząca do postępującego zwyrodnienia niektórych części układu nerwowego a także mięśnia sercowego. Po raz pierwszy opisana przez niemieckiego lekarza Nikolausa Friedreicha w 1863 roku.
 W
WChoroba Coriego – rzadka choroba genetyczna, dziedziczona w sposób autosomalny recesywny, spowodowana brakiem enzymu odszczepiającego glikogen (oligo-1,4:1,4-glukozotransferaza). Niedobór ten prowadzi do nadmiernego odkładania nieprawidłowego glikogenu w mięśniach, wątrobie, a także w sercu.
 W
WChoroba Gauchera – uwarunkowana genetycznie lizosomalna choroba spichrzeniowa o autosomalnym recesywnym sposobie dziedziczenia, spowodowana mutacją w genie GBA kodującym białko enzymu glukocerebrozydazy.
 W
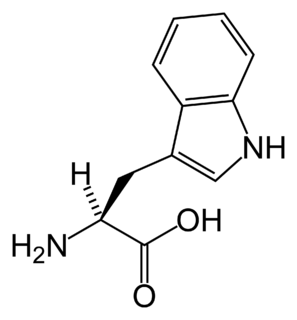
WChoroba Hartnupów – dziedziczone autosomalnie recesywnie zaburzenie transportu aminokwasów obojętnych, głównie tryptofanu, w świetle jelita i kanalikach nerkowych. Przyczyną choroby jest mutacja w genie SLC6A19 w locus 5p15. Choroba objawia się aminoacydurią i zwiększonym wydalaniem pochodnych indolu, np. indykanów. Częste są objawy przypominające pelagrę, wynikające z niedoboru tryptofanu niezbędnego do biosyntezy niacyny.
 W
WChoroba Taya-Sachsa – genetycznie uwarunkowana choroba z grupy chorób spichrzeniowych, polegająca na gromadzeniu się substancji tłuszczowej – gangliozydu GM2 w komórkach nerwowych mózgu. Nazwa choroby pochodzi od brytyjskiego okulisty Warrena Taya oraz amerykańskiego neurologa Bernarda Sachsa, który opisał zmiany komórkowe w tej chorobie oraz zauważył w 1887 roku częstsze występowanie choroby we wschodnioeuropejskiej populacji Żydów aszkenazyjskich. Eponim choroby Taya-Sachsa wprowadził w 1901 roku Henryk Higier.
 W
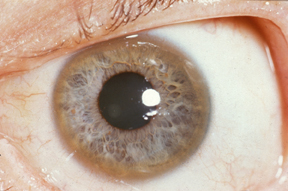
WChoroba Wilsona, zwyrodnienie soczewkowo-wątrobowe – uwarunkowane genetycznie zaburzenie metabolizmu miedzi w organizmie. Miedź, która zwykle jest wydzielana z żółcią, gromadzi się początkowo w wątrobie, prowadząc do jej uszkodzenia. Po przekroczeniu możliwości magazynowania tego pierwiastka, trafia on z krwią do innych tkanek, duże koncentracje osiągając zwłaszcza w mózgu, rogówkach oczu i nerkach. Objawy neurologiczne choroby Wilsona wynikają z uszkodzenia jąder podstawnych mózgu.
 W
WCystynuria – wrodzona, genetycznie uwarunkowana choroba metaboliczna o dziedziczeniu autosomalnym recesywnym, która charakteryzuje się wydalaniem zwiększonej ilości cystyny z moczem oraz upośledzeniem absorpcji z przewodu pokarmowego.
 W
WZespół Dubowitza – genetycznie uwarunkowany zespół wad wrodzonych charakteryzujący się niepełnosprawnością intelektualną, niedoborem wzrostu oraz zaburzeniami odporności predysponującymi do alergii, wyprysku, nowotworów układu krwiotwórczego oraz nerwiaka zarodkowego.
 W
WFenyloketonuria, oligofrenia fenylopirogronowa – wrodzona, uwarunkowana genetycznie enzymopatia prowadząca do gromadzenia się w organizmie nadmiaru aminokwasu fenyloalaniny i wynikających z niego toksycznych objawów chorobowych.U podłoża choroby leży mutacja genu odpowiedzialnego za aktywność enzymu hydroksylazy fenyloalaninowej (PAH), który jest niezbędny w metabolizmie fenyloalaniny.
 W
WGalaktozemia – choroba genetyczna, dziedziczona w sposób autosomalnie recesywny, wynikająca z gromadzenia się nadmiaru galaktozy i galaktozo-1-fosforanu z powodu braku enzymów odpowiadających za ich metabolizm. Galaktoza, obok glukozy, wchodzi w skład laktozy obecnej w mleku kobiecym i krowim.
 W
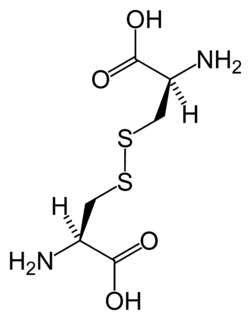
WHomocystynuria (ang. homocystinuria) – heterogenna etiologicznie, uwarunkowana genetycznie choroba metaboliczna, polegająca na nieprawidłowym metabolizmie aminokwasu metioniny. Homocystynuria charakteryzuje się podwyższonym poziomem homocysteiny w surowicy i w moczu. Najczęstszą postacią schorzenia jest homocystynuria spowodowana niedoborem i niską aktywnością enzymu syntazy β-cystationionowej (ang. cystathionine beta synthase deficiency), który katalizuje reakcję przekształcenia homocysteiny do cysteiny poprzez cystationinę. Reakcja katalizowana przez syntazę β-cystationionową wymaga udziału pirydoksyny (witaminy B6), dlatego w części przypadków homocystynurii obserwuje się poprawę po uzupełnieniu niedoboru pirydoksyny. Znanych jest dodatkowo przynajmniej siedem innych, znacznie rzadszych chorób genetycznych powodujących podobny blok metaboliczny; aby odróżnić niedobór CBS od tych rzadszych przyczyn używa się terminu klasycznej homocystynurii albo homocystynurii z powodu niedoboru syntazy β-cystationionowej. Dziedziczenie choroby jest autosomalne recesywne, obydwa allele genu CBS muszą być zmutowane, aby homocystynuria ujawniła się klinicznie. Heterozygotyczni nosiciele mutacji w jednym allelu genu CBS nie chorują. Ryzyko urodzenia kolejnego dziecka z homocystynurią wynosi 25%.
 W
WZespół Johanson-Blizzarda – genetycznie uwarunkowany zespół wad wrodzonych charakteryzujący się niedoczynnością zewnątrzwydzielniczą trzustki, hipoplazją skrzydełek nosa, hipodoncją, niedosłuchem odbiorczym oraz niepełnosprawnością intelektualną.
 W
WKarłowatość typu Larona – rzadka choroba uwarunkowana genetycznie, wynikająca z defektu odpowiedzi receptorów obwodowych dla hormonu wzrostu. Jest dziedziczona w sposób autosomalny recesywny. Charakteryzuje się dużym wydzielaniem somatotropiny i brakiem wydzielania somatomedyny C. Receptory nie odpowiadają w tym przypadku na GH. Cechą charakterystyczną zespołu oprócz karłowatości jest otyłość centralna, hiperlipidemia, insulinooporność i małe prącie.
 W
WLeprechaunizm, zespół Donohue, krasnoludkowatość – rzadka, genetycznie uwarunkowana choroba o dziedziczeniu autosomalnym recesywnym. U podłoża tego schorzenia leży mutacja genu dla receptora insulinowego (INSR) na krótkim ramieniu chromosomu 19, co prowadzi do zmiany jego funkcji receptora i najcięższej postaci insulinoooporności.
 W
WMukowiscydoza – wrodzona choroba uwarunkowana genetycznie polegająca na zaburzeniu wydzielania przez gruczoły zewnątrzwydzielnicze.
 W
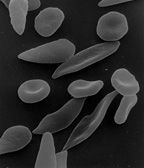
WNiedokrwistość sierpowata, anemia sierpowata (łac. anaemia drepanocytica, ang. sickle cell anemia) – rodzaj wrodzonej niedokrwistości spowodowanej nieprawidłową budową hemoglobiny. Mutacja punktowa w genie łańcucha β (HBB) hemoglobiny powoduje zmianę pojedynczego aminokwasu w sekwencji białka (z kwasu glutaminowego na walinę, w pozycji 6 od końca NH2). Hemoglobinę z tak zmienioną, nieprawidłową strukturą I-rzędową określa się jako hemoglobinę S (HbS) w przeciwieństwie do normalnej, występującej u dorosłych hemoglobiny A (HbA). Hemoglobina S charakteryzuje się zmienionymi w porównaniu z hemoglobiną A własnościami fizykochemicznymi.
 W
WPierwotna dyskineza rzęsek – rzadka, genetycznie uwarunkowana choroba dziedziczona w sposób autosomalny recesywny, w której objawy chorobowe wywołane są przez nieprawidłową budowę rzęsek pokrywających nabłonki urzęsione organizmu człowieka. PCD należy, razem z innymi chorobami spowodowanymi dysfunkcją rzęsek, do grupy ciliopatii. Najpoważniejsze objawy wynikają z upośledzenia funkcji nabłonka oddechowego górnych i dolnych dróg oddechowych. Choroba ta jest również przyczyną bezpłodności u mężczyzn, ponieważ defekt budowy dotyczy również witek plemników.
 W
WPłód arlekin – rzadka choroba genetyczna, o dziedziczeniu autosomalnym recesywnym. Rybia łuska arlekinowa należy do genodermatoz i razem z kilkoma podobnymi schorzeniami do grupy autosomalnie recesywnie dziedziczonej wrodzonej rybiej łuski. Obraz kliniczny charakteryzuje się pokrywającymi całą powierzchnię skóry łuskami hiperkeratotycznego naskórka o kształcie rombów i wielokątów, o ułożeniu przywodzącym na myśl kostium arlekina – stąd nazwa jednostki chorobowej. Ponadto stwierdza się niską masę urodzeniową, erytrodermię, wywinięcie warg (eclabium) i powiek (ektropion). Z powodu utraty wody i sepsy oraz nieprawidłowej termoregulacji noworodek umiera zwykle w ciągu tygodnia. Przynajmniej część przypadków wiąże się z mutacjami genu ABCA12 na chromosomie 2.
 W
WZespół Rothmunda-Thompsona – rzadki zespół wad wrodzonych charakteryzujący się występowaniem:niskiego wzrostu, zmian skórnych, skóra marmurkowata, teleangiektazje, zmiany pigmentacji, defektów paznokci, zębów, wrodzonej zaćmy, łysienia, wrodzonych zaburzeń kostnych, zwiększonego ryzyka zachorowania na kostniakomięsaka.
 W
WSkóra pergaminowa – bardzo rzadkie schorzenie dziedziczone autosomalnie recesywnie spowodowane genetycznym defektem polimerazy DNA β, skutkiem czego dochodzi do nieodwracalnych uszkodzeń DNA w komórkach skóry narażonych na działanie promieni UV.
 W
WTalasemia – ilościowe zaburzenia syntezy hemoglobiny, spowodowane wrodzonym defektem biosyntezy łańcuchów globiny. Najczęściej zaburzenia dotyczą ekspresji alfa-globiny (alfa-talasemia) lub beta-globiny (beta-talasemia), choć istnieją także talasemie związane z obniżoną syntezą innych globin np. delta-globiny, gamma-globiny. Zaburzenia w biosyntezie globin spowodowane są mutacjami w kodujących je genach lub ich elementach regulatorowych.
 W
WWrodzony przerost nadnerczy – grupa chorób charakteryzujących się nadmiernym wydzielaniem androgenów nadnerczowych z niedoborem kortyzolu wskutek genetycznie uwarunkowanego braku enzymów szlaku biosyntezy hormonów nadnerczy, przede wszystkim 21-hydroksylazy i 11β-hydroksylazy.
 W
WZespół Blooma – rzadka choroba uwarunkowana genetycznie, charakteryzująca się zwiększoną predyspozycją do uszkodzeń chromosomów. Chorobę odkrył i opisał w 1954 roku David Bloom.