 W
WAcyduria mewalonianowa – choroba metaboliczna uwarunkowana genetycznie. Spowodowana jest niedoborem enzymu kinazy mewalonianowej. Objawia się opóźnieniem umysłowym, postępującą ataksją, przełomami gorączkowymi z biegunką i wymiotami, wysypką na skórze, hepatosplenomegalią, cechami dysmorficznymi twarzy, nieprawidłowym rozwojem i wzrostem.
 W
WAlfa-mannozydoza – uwarunkowana genetycznie choroba z grupy lizosomalnych chorób spichrzeniowych, charakteryzująca się pierwotnym niedoborem odporności, nieprawidłowościami szkieletu i cechami dysmorficznymi twarzy, upośledzeniem słuchu i opóźnieniem umysłowym. Chorobę opisał Ockerman w 1967 roku.
 W
WChoroba Andersen, glikogenoza typu IV, amylopektynoza – rzadka choroba genetyczna, dziedziczona autosomalnie recesywnie, polegająca na zaburzeniu spichrzania glikogenu.
 W
WChoroba Coriego – rzadka choroba genetyczna, dziedziczona w sposób autosomalny recesywny, spowodowana brakiem enzymu odszczepiającego glikogen (oligo-1,4:1,4-glukozotransferaza). Niedobór ten prowadzi do nadmiernego odkładania nieprawidłowego glikogenu w mięśniach, wątrobie, a także w sercu.
 W
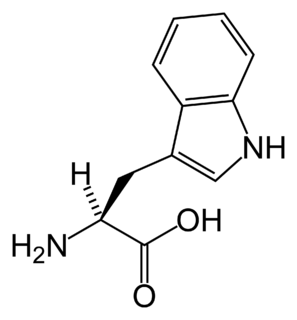
WChoroba Hartnupów – dziedziczone autosomalnie recesywnie zaburzenie transportu aminokwasów obojętnych, głównie tryptofanu, w świetle jelita i kanalikach nerkowych. Przyczyną choroby jest mutacja w genie SLC6A19 w locus 5p15. Choroba objawia się aminoacydurią i zwiększonym wydalaniem pochodnych indolu, np. indykanów. Częste są objawy przypominające pelagrę, wynikające z niedoboru tryptofanu niezbędnego do biosyntezy niacyny.
WChoroba Hersa, glikogenoza typu VI, GSD VI – rzadka choroba genetyczna, dziedziczona w sposób autosomalny recesywny spowodowana brakiem fosforylazy glikogenowej (wątrobowej). Niedobór ten prowadzi do nadmiernego odkładania glikogenu w wątrobie.
 W
WChoroba tangierska – choroba genetyczna, dziedziczona autosomalnie recesywnie. Charakteryzuje się znacznym obniżeniem poziomu HDL we krwi. Jest spowodowana mutacją w położonym na chromosomie 9 genie ABCA1, kodującym białko transportujące cholesterol i fosfolipidy z komórki. Została pierwotnie odkryta na wyspie Tangier w pobliżu wybrzeży Wirginii.
 W
WChoroba Taya-Sachsa – genetycznie uwarunkowana choroba z grupy chorób spichrzeniowych, polegająca na gromadzeniu się substancji tłuszczowej – gangliozydu GM2 w komórkach nerwowych mózgu. Nazwa choroby pochodzi od brytyjskiego okulisty Warrena Taya oraz amerykańskiego neurologa Bernarda Sachsa, który opisał zmiany komórkowe w tej chorobie oraz zauważył w 1887 roku częstsze występowanie choroby we wschodnioeuropejskiej populacji Żydów aszkenazyjskich. Eponim choroby Taya-Sachsa wprowadził w 1901 roku Henryk Higier.
WChoroby spichrzeniowe glikogenu (glikogenozy) – genetycznie uwarunkowane choroby metaboliczne, prowadzące do nieprawidłowego magazynowania glikogenu w wątrobie, nerkach i mięśniach.
 W
WCukrzyca – grupa chorób metabolicznych charakteryzująca się hiperglikemią wynikającą z defektu produkcji lub działania insuliny wydzielanej przez komórki beta trzustki. Przewlekła hiperglikemia wiąże się z uszkodzeniem, zaburzeniem czynności i niewydolnością różnych narządów, szczególnie oczu, nerek, nerwów, serca i naczyń krwionośnych. Ze względu na przyczynę i przebieg choroby, można wyróżnić: cukrzycę typu 1, typu 2, cukrzycę ciężarnych i inne, rzadziej występujące typy.
WCukrzyca ciężarnych, cukrzyca ciążowa – każdy stan hiperglikemiczny pojawiające się u zdrowych dotąd kobiet w ciąży. Stanowi ono zagrożenie dla płodu, a ponadto u 30-45% kobiet, u których stwierdzano cukrzycę ciężarnych, w ciągu najbliższych 15 lat rozwija się cukrzyca typu 2.
WCukrzyca typu 1, in. cukrzyca insulinozależna – jedna z etiopatogenetycznych postaci cukrzycy. U jej podłoża choroby leży przewlekły, autoimmunologiczny proces chorobowy prowadzący do powolnego zniszczenia produkujących insulinę komórek β wysp trzustkowych i w następstwie tego do utraty zdolności jej wydzielania. Choroba leczona jest, poprzez podawanie przez cały okres życia pacjenta, insuliny o krótkim czasie działania do spożywanych w ciągu dnia posiłków, a także insulinę o długo działającym, bezszczytowym analogu – tzw. bazę, która podawana jest raz na dobę. Leczenie wymaga od pacjenta kilkukrotnych podskórnych wstrzyknięć leku w roztworze, a także systematycznego, wielokrotnego oznaczania poziomu glukozy we krwi. Jest to model funkcjonalnej intensywnej insulinoterapii, niezbędnej do normalnego funkcjonowania chorego.
WCukrzyca typu MODY – grupa rzadkich i uwarunkowanych genetycznie postaci cukrzycy, których objawy pojawiają się u osób w wieku 15-35 lat, a ich przebieg kliniczny jest zbliżony do cukrzycy typu 2. W przeciwieństwie do typu 1, który zawsze wymaga przyjmowania insuliny, chorzy z MODY często mogą przyjmować leki doustne. Stanowią 1–5% wszystkich przypadków cukrzycy.
 W
WDziedziczna koproporfiria – rzadka choroba genetyczna, należąca do grupy ostrych porfirii wątrobowych. Objawem odróżniającym ją od innych jest obecność koproporfirynogenu III w kale i moczu, przekraczająca nawet 200-krotnie ilość spotykaną o osób zdrowych.
 W
WFenyloketonuria, oligofrenia fenylopirogronowa – wrodzona, uwarunkowana genetycznie enzymopatia prowadząca do gromadzenia się w organizmie nadmiaru aminokwasu fenyloalaniny i wynikających z niego toksycznych objawów chorobowych.U podłoża choroby leży mutacja genu odpowiedzialnego za aktywność enzymu hydroksylazy fenyloalaninowej (PAH), który jest niezbędny w metabolizmie fenyloalaniny.
 W
WHemochromatoza dziedziczna, inaczej wrodzona lub pierwotna, dawniej stosowane nazwy to: cukrzyca brunatna i cukrzyca brązowa – genetycznie uwarunkowana choroba metabolizmu żelaza, w której dochodzi do nadmiernego wchłaniania tego pierwiastka z pożywienia i nadmiernego gromadzenia w tkankach, co prowadzi do objawów chorobowych.
 W
WHomocystynuria (ang. homocystinuria) – heterogenna etiologicznie, uwarunkowana genetycznie choroba metaboliczna, polegająca na nieprawidłowym metabolizmie aminokwasu metioniny. Homocystynuria charakteryzuje się podwyższonym poziomem homocysteiny w surowicy i w moczu. Najczęstszą postacią schorzenia jest homocystynuria spowodowana niedoborem i niską aktywnością enzymu syntazy β-cystationionowej (ang. cystathionine beta synthase deficiency), który katalizuje reakcję przekształcenia homocysteiny do cysteiny poprzez cystationinę. Reakcja katalizowana przez syntazę β-cystationionową wymaga udziału pirydoksyny (witaminy B6), dlatego w części przypadków homocystynurii obserwuje się poprawę po uzupełnieniu niedoboru pirydoksyny. Znanych jest dodatkowo przynajmniej siedem innych, znacznie rzadszych chorób genetycznych powodujących podobny blok metaboliczny; aby odróżnić niedobór CBS od tych rzadszych przyczyn używa się terminu klasycznej homocystynurii albo homocystynurii z powodu niedoboru syntazy β-cystationionowej. Dziedziczenie choroby jest autosomalne recesywne, obydwa allele genu CBS muszą być zmutowane, aby homocystynuria ujawniła się klinicznie. Heterozygotyczni nosiciele mutacji w jednym allelu genu CBS nie chorują. Ryzyko urodzenia kolejnego dziecka z homocystynurią wynosi 25%.
 W
WJamajska choroba wymiotna – zespół objawów zatrucia niedojrzałymi owocami drzewa ackee, zawierającymi hipoglicynę A i B. Metabolit hipoglicyny A – kwas metylenocyklopropylooctowy (MCPA) – inaktywuje enzym dehydrogenazę krótko- i średniołańcuchowych acylo-CoA, powodując zahamowanie β-oksydacji. Choroba objawia się ostrymi objawami ze strony przewodu pokarmowego i hipoglikemią, w ciężkich przypadkach może dojść do depresji ośrodkowego układu nerwowego, a u pacjenta z cechą sierpowatokrwinkowości opisano piorunującą niewydolność wątroby, która wymagała przeszczepu.
 W
WKępki żółte, in. żółtaki – żółtawe zmiany grudkowe umiejscowione najczęściej w okolicach powiek (xanthelasma), związane z odkładaniem cholesterolu w skórze.
 W
WKwashiorkor – dotycząca najczęściej dzieci w ubogich krajach choroba powodowana tak niedoborem ilościowym, jak i jakościowym pożywienia. Niedobory żywieniowe zaburzają syntezę enzymów, niedostateczna podaż aminokwasów prowadzi do zmian funkcji, a potem również struktury narządów wewnętrznych, wtórnie – także do zaburzeń gospodarki wodno-elektrolitowej i układu odpornościowego, dochodzi do zakażeń, w tym na przykład rzadko spotykanego u osób prawidłowo żywionych raka wodnego.
 W
WNiedobór syntazy metioninowej, niedobór metylotransferazy homocysteinowej, zespół Arakawy II (ang. methionine synthase deficiency, tetrahydrofolate-methyltransferase deficiency syndrome, N5-methylhomocysteine transferase deficiency, Arakawa syndrome II) to genetycznie uwarunkowana choroba metaboliczna wynikająca z niedoboru enzymu metylotransferazy homocysteinowej. Dotknięte nią osoby nie mogą właściwie metabolizować metylokobalaminy, jednej z postaci witaminy B12.
 W
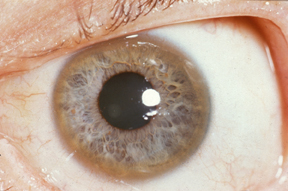
WPierścień Kaysera i Fleischera – objaw choroby Wilsona występujący w postaci złocistego lub złocisto-brązowego przebarwienia rogówki, szczególnie dobrze widoczny u osób niebieskookich – u innych konieczne jest badanie lampą szczelinową. Powstaje on przez gromadzenie się w błonie Descemeta rogówki osadów miedzi, co nie upośledza wzroku, jest jednak sygnałem uwolnienia się tego pierwiastka z wątroby i uszkodzenia mózgu. Niekiedy (rzadko) pierścieniowi Kaysera i Fleischera towarzyszy tak zwana zaćma słonecznikowa. Brak pierścienia w badaniu nie wyklucza choroby Wilsona.
 W
WProtoporfiria erytropoetyczna – wrodzone zaburzenie metabolizmu hemu (porfiria) spowodowane w większości przypadków niedoborem enzymu ferrochelatazy i podwyższonymi poziomami protoporfiryn w tkankach. Jest stosunkowo łagodną postacią porifrii, jednak wiąże się z nasilonymi dolegliwościami bólowymi. Zagrażającym życiu powikłaniem jest ostra niewydolność wątroby. Chorobę opisał Ian Allingham Magnus ze współpracownikami z londyńskiego St John's Institute of Dermatology w 1961 roku.
 W
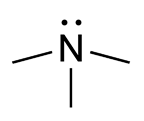
WTrimetyloaminuria (TMAU), znana też pod angielską nazwą fish odor syndrome – rzadkie zaburzenie metaboliczne powodujące niedobór w produkcji enzymu FMO3.
 W
WZespół chylomikronemii – stosunkowo rzadka choroba z grupy zaburzeń lipidowych. Istotą choroby jest stała obecność chylomikronów w osoczu. Klinicznie może się objawiać napadowymi ostrymi bólami brzucha lub ostrym zapaleniem trzustki.
 W
WZespół Criglera-Najjara – rzadka choroba uwarunkowana genetycznie, będąca przyczyną ciężkiej żółtaczki.