 W
WAkromegalia – choroba spowodowana nadmiernym wydzielaniem hormonu wzrostu (somatotropiny) przez hormonalnie czynnego gruczolaka komórek kwasochłonnych przedniego płata przysadki mózgowej. Akromegalia występuje u osób dorosłych, u których skończony został proces wzrastania kości, a nasady kości długich uległy mineralizacji i zrośnięciu. U dzieci i młodzieży nadmiar hormonu wzrostu powoduje gigantyzm, odróżniający się od akromegalii nadmiernym wzrostem kości długich.
 W
WChoroba Addisona, pierwotna niedoczynność kory nadnerczy, dawna nazwa cisawica – schorzenie układu dokrewnego, zespół objawów spowodowanych przewlekłym niedoborem hormonów produkowanych przez korę nadnerczy.
 W
WCukrzyca – grupa chorób metabolicznych charakteryzująca się hiperglikemią wynikającą z defektu produkcji lub działania insuliny wydzielanej przez komórki beta trzustki. Przewlekła hiperglikemia wiąże się z uszkodzeniem, zaburzeniem czynności i niewydolnością różnych narządów, szczególnie oczu, nerek, nerwów, serca i naczyń krwionośnych. Ze względu na przyczynę i przebieg choroby, można wyróżnić: cukrzycę typu 1, typu 2, cukrzycę ciężarnych i inne, rzadziej występujące typy.
WCukrzyca typu 1, in. cukrzyca insulinozależna – jedna z etiopatogenetycznych postaci cukrzycy. U jej podłoża choroby leży przewlekły, autoimmunologiczny proces chorobowy prowadzący do powolnego zniszczenia produkujących insulinę komórek β wysp trzustkowych i w następstwie tego do utraty zdolności jej wydzielania. Choroba leczona jest, poprzez podawanie przez cały okres życia pacjenta, insuliny o krótkim czasie działania do spożywanych w ciągu dnia posiłków, a także insulinę o długo działającym, bezszczytowym analogu – tzw. bazę, która podawana jest raz na dobę. Leczenie wymaga od pacjenta kilkukrotnych podskórnych wstrzyknięć leku w roztworze, a także systematycznego, wielokrotnego oznaczania poziomu glukozy we krwi. Jest to model funkcjonalnej intensywnej insulinoterapii, niezbędnej do normalnego funkcjonowania chorego.
WCukrzyca typu MODY – grupa rzadkich i uwarunkowanych genetycznie postaci cukrzycy, których objawy pojawiają się u osób w wieku 15-35 lat, a ich przebieg kliniczny jest zbliżony do cukrzycy typu 2. W przeciwieństwie do typu 1, który zawsze wymaga przyjmowania insuliny, chorzy z MODY często mogą przyjmować leki doustne. Stanowią 1–5% wszystkich przypadków cukrzycy.
 W
WGuz gastrynowy – guz neuroendokrynny, drugi co do częstości występowania typ wyspiaka trzustki po insulinoma. Zapadalność wynosi 1:1 000 000/rok. Lokalizuje się głównie w głowie trzustki i dwunastnicy. Wywodzi się z komórki G, Zwykle nie osiąga rozmiarów powyżej 1 cm. Złośliwość ocenia się na 60% przypadków. Jest najczęstszym guzem neuroendokrynnym w zespole MEN1. Guzy gastrynowe mogą wydzielać oprócz gastryny również ACTH.
 W
WGigantyzm – ogromne rozmiary ciała na skutek nadmiernego wzrostu całego kośćca i masy tkanek, mocno przekraczającego normy dla danej rasy.
 W
WGruczolak przysadki – najczęściej występujący guz przysadki, stanowiący około 10% wszystkich guzów wewnątrzczaszkowych. Jest on najczęstszą przyczyną zaburzeń osi podwzgórze-przysadka. Może być bezobjawowy; gdy ujawnia się klinicznie, obraz choroby jest zróżnicowany w zależności od czynności hormonalnej guza i jego wielkości.
 W
WGuz insulinowy, wyspiak, insulinoma – nowotwór wywodzący się z komórek B wysp trzustkowych, wytwarzający insulinę, w związku z czym głównym jego objawem jest hipoglikemia.
 W
WHiperprolaktynemia – podwyższone stężenie prolaktyny we krwi. Osiowe objawy hiperprolaktynemii dają obraz wtórnego hipogonadyzmu hipogonadotropowego.
 W
WMikropenis – wada rozwojowa zewnętrznych męskich narządów płciowych polegająca na wykształceniu u osoby z kariotypem 46XY prawidłowego prącia, o prawidłowej funkcji, lecz o długości w stanie wzwodu mniejszej o co najmniej 2,5 odchylenia standardowego od średniej długości prącia, czyli mniejszy niż około 7 cm. Przyczyną jest niedobór androgenów w drugiej fazie rozwoju narządów płciowych. Leczenie polega na hormonoterapii.
 W
WMnoga gruczolakowatość wewnątrzwydzielnicza typu 2 – rzadka choroba genetyczna objawiająca się predyspozycją do raka rdzeniastego tarczycy, guza chromochłonnego nadnerczy i nadczynności przytarczyc, a także innych rzadszych guzów i wad wrodzonych. Chorobę wywołują mutacje w protoonkogenie RET; podobna nieco w obrazie klinicznym mnoga gruczolakowatość wewnątrzwydzielnicza typu 1 ma inną etiologię.
 W
WMoczówka prosta – choroba, w której na skutek niezdolności do zagęszczania moczu dochodzi do wydalania bardzo dużych ilości rozcieńczonego moczu. Spowodowana jest bezwzględnym lub względnym niedoborem wazopresyny, bądź niewrażliwością tkanek na jej działanie. Objawia się przymusem picia dużej ilości płynów (polidypsja), postępującym odwodnieniem, upośledzeniem krążenia i ciężką hipernatremią. Ilość produkowanego moczu wynosić może od 5 do nawet 25 litrów dziennie.
 W
WZespół Morgagniego-Stewarta-Morela – rzadka choroba genetyczna, która charakteryzuje się trzema głównymi objawami: 1) przerostem wewnętrznej powierzchni kości czołowej, 2) otyłością i 3) wirylizmem i hirsutyzmem.
 W
WNadczynność przytarczyc – nadaktywność przytarczyc w wyniku której dochodzi do nadmiernej produkcji parathormonu (PTH). Parathormon jest hormonem regulującym poziom wapnia i fosforanów. Nadczynność przytarczyc dzieli się na:pierwotną nadczynność przytarczyc wtórną nadczynność przytarczyc
 W
WRak nadnercza – rzadki nowotwór złośliwy, wywodzący się z kory nadnerczy.
 W
WRakowiak – nowotwór wywodzący się z jelitowych komórek neuroendokrynnych.
 W
WRozrost mikroguzkowy nadnerczy – rzadka choroba kory nadnerczy, występująca sporadycznie lub dziedzicznie. Obok rozrostu nadnerczy u chorych mogą występować śluzaki i plamista pigmentacja skóry. Dziedziczny rozrost mikroguzkowy nadnerczy znany jest także jako zespół Carneya od nazwiska amerykańskiego patologa J. Aidana Carneya, który opisał go jako pierwszy w artykule na łamach New England Journal of Medicine. Należy rozróżnić pierwotny pigmentowany rozrost drobnoguzkowy nadnerczy od triady Carneya, opisanej przez tego samego badacza w 1983 roku, na którą składają się mięsak podścieliskowy żołądka, chrzęstniak płuca i pozanadnerczowy przyzwojak.
 W
WRzekoma niedoczynność przytarczyc – grupa genetycznie uwarunkowanych chorób metabolicznych, charakteryzujących się opornością tkanek docelowych na parathormon. Wyróżniono cztery typy choroby: Ia, Ib, Ic i II. W dwóch z nich stwierdzono mutację genu receptora dla PTH (PTHrP). W typie Ia stwierdza się ponadto oporność receptorów dla TSH, glukagonu i gonadotropin, w typie II reakcja receptorów dla TSH, glukagonu i gonadotropin jest prawidłowa. Obraz kliniczny chorób jest zróżnicowany; obok hipokalcemii i hiperfosfatemii różnego stopnia, mogą występować niedoczynność tarczycy i gonad, niski wzrost, okrągła twarz, otyłość, charakterystycznie skrócone kości śródręcza i śródstopia, zwapnienia podskórne. Typowy dla choroby fenotyp może występować u członków rodziny pacjenta, mimo prawidłowego profilu hormonów; stan taki nazywa się rzekomo-rzekomą niedoczynnością przytarczyc (pseudopseudohypoparathyroidismus).
 W
WUdar przysadki – choroba ośrodkowego układu nerwowego polegająca na zawale lub krwawieniu do przysadki mózgowej, często związanymi z obecnym gruczolakiem przysadki.
 W
WWrodzony przerost nadnerczy – grupa chorób charakteryzujących się nadmiernym wydzielaniem androgenów nadnerczowych z niedoborem kortyzolu wskutek genetycznie uwarunkowanego braku enzymów szlaku biosyntezy hormonów nadnerczy, przede wszystkim 21-hydroksylazy i 11β-hydroksylazy.
 W
WZespół Allgrove’a – rzadki, uwarunkowany genetycznie zespół o dziedziczeniu autosomalnym recesywnym. Na obraz kliniczny zespołu składa się triada:choroba Addisona brak wydzielania łez (alakrimia) achalazja przełyku.
 W
WZespół Conna, inaczej pierwotny hiperaldosteronizm – zespół chorobowy wywołany zwiększonym wytwarzaniem aldosteronu z jednoczesnym zahamowaniem aktywności reninowej osocza. Stwierdzany jest najczęściej u osób między 3. a 5. dekadą życia.
 W
WZespół Cushinga – zespół objawów chorobowych związanych z występowaniem podwyższonego poziomu kortyzolu w surowicy krwi. Najczęstszą przyczyną występowania zespołu Cushinga jest długotrwałe podawanie glikokortykosterydów w leczeniu innych chorób.
 W
WZespół Kallmanna – grupa uwarunkowanych genetycznie chorób endokrynologicznych, w których obrazie klinicznym występuje zaburzenie węchu i hipogonadyzm hipogonadotropowy.
 W
WZespół pustego siodła – zespół objawów spowodowanych patologią przysadki, polegającą na przewlekłym wpuklaniu się przestrzeni podpajęczynówkowej w obręb siodła tureckiego.
WZespół Schwartza-Barttera – choroba wywołana nadmiernym uwalnianiem hormonu antydiuretycznego (ADH) przez tylny płat przysadki mózgowej lub wydzielaniem ektopowym, hiponatremią i hipoosmolarnością płynów ustrojowych.
 W
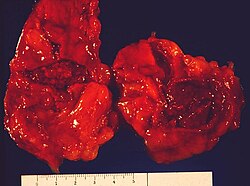
WZespół Waterhouse’a-Friderichsena – zespół objawów spowodowanych masywnym, zazwyczaj obustronnym krwotokiem do nadnerczy w przebiegu posocznicy, najczęściej wywołanej przez Neisseria meningitidis. Choroba ta przebiega piorunująco i potrafi doprowadzić do śmierci w ciągu kilku godzin.
 W
WZespół wielotorbielowatych jajników – zaburzenie endokrynne, które dotyka 10–15% kobiet w okresie reprodukcyjnym. Występuje wśród wszystkich ras i narodowości i jest najczęstszym zaburzeniem hormonalnym u kobiet w wieku rozrodczym, a także najczęstszą przyczyną niepłodności. Objawy oraz stopień ich nasilenia różnią się bardzo pomiędzy kobietami. Przyczyny zespołu są nieznane, ale wiadomo, że insulinooporność jest silnie powiązana z PCOS.
 W
WZłamanie rzekome in. pseudozłamanie, zespół Milkmana, strefa Loosera-Milkmana, Looserowskie strefy przebudowy, strefa Loosera – charakterystyczny dla osteomalacji obraz zmian radiologicznych w kościach u człowieka.